

Link to our crystal structures in the Cambridge Structural Database
10-Phenyl-10H-phenoxazine-4,6-diol Tetrahydrofuran Monosolvate

In the crystalline state of the title compound, 10-Phenyl-10H-phenoxazine-4,6-diol, hydrogen-bonding interactions between hydroxyl groups on a phenoxazine backbone and tetrahydrofuran solvent are observed that suggest the ability for this compound to act as a chelating ligand. The donor-acceptor distances for this hydrogen-bonding are 2.7729 (15) Å for O2—O4 and 2.7447 (15)
Å for O3—O4. The three-ring structure of the phenoxazine bends out of planarity by 18.92
(3)°, with N1 and O1 forming the line of flexion, as computed using mean planes that encompass each half of the three-ring structure. A π-π stacking arrangement exists between inversion-related (1-x, 1-y, 1-z) molecules with a centroid-to-centroid distance of 3.6355 (11)
Å. In the disordered tetrahydrofuran solvate, all atoms except oxygen were modeled over two positions, with occupancies varying freely and arriving at 0.511 (8) and 0.489 (8).
- Whalen, A. C.; Hernandez Brito, C.; Choi, K. H.; Warner, E. J. T.; Thole, D. A.; Gau, M. R.; Carroll, P. J.; Anstey, M. R. “10-Phenyl-10H-phenoxazine-4,6-diol tetrahydrofuran monosolvate.” IUCrData 2020, 5, x201276.
2,2′-Oxybis(1,3-bis(4-methoxyphenyl)-2,3-dihydro-1H-benzo[d][1,3,2]diazaborole)
![The solid state molecular structure of 2,2'-Oxybis(1,3-bis(4-methoxyphenyl)-2,3-dihydro-1H-benzo[d][1,3,2]diazaborole) obtained from an X-ray crystal structure analysis.](http://www.mitchanstey.org/wp-content/uploads/2020/09/obdr-019a-794x1024.png)
In the title compound, C40H36B2N4O5, the B—O—B bond angle is 132.75 (13) and the dihedral angle between the benzodiazborole rings is 73.02 (5)°. In the crystal, weak C—H···O interactions link the molecules.
- Mallard, H. H.; Kennedy, N. D.; Rudman, N. A.; Greenwood, A. M.; Nicoleau, J.; Angle, C. E.; Torquato, N. A.; Gau, M. R.; Carroll, P. J.; Anstey, M. R. “2,2’-Oxybis[1,3-bis(4-methoxyphenyl)-2,3-dihydro-1H-benzo[d][1,3,2]diazaborole].” IUCrData, 2020, 5, x201248.
#DavidsonTrue: Transitioning to Remote Teaching while Maintaining Our Values as a Liberal Arts College during the COVID-19 Pandemic

The coronavirus 2019 (COVID-19) outbreak in March led Davidson College to move from face-to-face classes and laboratories to mostly synchronous Zoom meetings. Prior to COVID-19, the majority of our faculty and students had little experience with remote instruction. With only 5 days to develop a plan, we revisited our individual and department learning goals and worked collectively to help each other redesign and redeploy our courses. In this reflective piece, we provide examples of how each member of our department collaborated with our students to ensure a relatively smooth transition to remote teaching across our entire curriculum while maintaining inclusive excellence. Specific strategies for adapting class meetings, assignments, assessments, additional support, and labs are provided along with select examples. Common themes across the curriculum included increased flexibility, the desire to maintain community, and the need for additional academic, technical, and emotional support. We hope our reflections will be helpful to our chemistry colleagues as we move into the uncertainty of the fall semester.
- Anstey, M. R.; Blauch, D. N; Carroll, F. A.; Gorensek-Benitez, A. H.; Hauser, C. D.; Key, H. M.; Myers, J. K.; Stevens, E. P.; Striplin, D. R.; Holck, H. W.; Montero-Lopez, L.; Snyder, N. L. Journal of Chemical Education 2020, 97, 2800-2805.
Crystal Structures of trans-Acetyldicarbonyl(η5-cyclopentadienyl)(1,3,5-triaza-7-phosphaadamantane)molybdenum(II) and trans-Acetyldicarbonyl(η5-cyclopentadienyl)(3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane)molybdenum(II)

The title compounds, [Mo(C5H5)(COCH3)(C6H12N3P)(CO)2], (1), and [Mo(C5H5)(COCH3)(C9H16N3O2P)(C6H5)2))(CO)2], (2), have been prepared by phosphine-induced migratory insertion from [Mo(C5H5)(CO)3(CH3)]. The molecular structures of these complexes are quite similar, exhibiting a four-legged piano-stool geometry with trans-disposed carbonyl ligands. The extended structures of complexes (1) and (2) differ substantially. For complex (1), the molybdenum acetyl unit plays a dominant role in the organization of the extended structure, joining the molecules into centrosymmetrical dimers through C—H—O interactions with a cyclopentadienyl ligand of a neighboring molecule, and these dimers are linked into layers parallel to (100) by C—H—O interactions between the molybdenum acetyl and the cyclopentadienyl ligand of another neighbor. The extended structure of (2) is dominated by C—H—O interactions involving the carbonyl groups of the acetamide groups of the DAPTA ligand, which join the molecules into centrosymmetrical dimers and link them into chains along [010]. Additional C—H—O interactions between the molybdenum acetyl oxygen atom and an acetamide methyl group join the chains into layers parallel to (101).
- Anstey, M. R.; Bost, J. L.; Grumman, A. S.; Kennedy, N. D.; Whited, M. T. Acta Crystallographica Section E 2020, 76, 547-551.
Desymmetrized Hexasubstituted [3]radialene Anions as Aqueous Organic Catholytes for Redox Flow Batteries

Negatively substituted trimethylenecyclopropane dianions, a subclass of hexasubstituted [3]radialenes, are candidates for use as active species in redox flow batteries (RFBs) due to their stability in water, reversible electrochemistry, and their tailorable synthesis. Hexacyano[3]radialene disodium is investigated as a pH 7 aqueous organic catholyte. The dianion and radical anion are stable in air and aqueous solutions at neutral pH. Systematic introduction of asymmetry via step-wise synthesis leads to enhanced solubility and higher capacity retention during galvanostatic cycling. An aqueous flow cell comprising a disester-tetracyano[3]radialene catholyte, sulfonated-methyl viologen as the anolyte, and a cation exchange membrane provides an operating Vcell = 0.9 V, 99.609 % coulombic efficiency, and minimum capacity fade over 50 cycles.
- Turner, N. A.; Freeman, M. B.; Pratt, H. D., III; Crockett, A. E.; Jones, D. S.; Anstey, M. R.; Anderson, T. M.; Bejger, C. M. Chemical Communications 2020, 56, 2739-2742.
A Method for Calibrating the Relative Gamma-Ray Light Yield of Plastic Scintillators
Currently, we are investigating the inclusion of organotin compounds in new polystyrene scintillator materials to improve full gamma-ray energy sensitivity. Accurate calibration of the relative light yield from the newly developed scintillators is crucial to assess merits of compounds and chemical processes used in the scintillators’ development. In this study, we present a spectral gain matching of measured and simulated spectra, using a spectrum rebinning technique, to determine the Compton edge in a measured Compton continuum for accurate relative light yield calibration. The Compton edges determined using this technique were found to be within 1.2% of their theoretical estimates.
- Mengesha, W.; Feng, P. L.; Cordaro, J. G.; Anstey, M. R.; Myllenbeck, N. R.; Throckmorton, D. J. Review of Scientific Instruments 2017, 88, 035108.
Control of Clustering Behavior in Anionic Cerium(III) Corrole Complexes: from Oligomers to Monomers

The first synthesis of anion-capped cerium corrole complexes is reported. Unusual clustering of the lanthanide corrole units has been found, and the degree of aggregation can be controlled by the choice of the capping ligand. A polymeric structure is formed using sodium cyclopentadienide (NaCp) and a dimeric structure is formed when potassium tris(pyrazolyl)borate (KTp) is used. Encapsulation of the counter-cation leads to the isolation of the monomeric structures.
- Armstrong, K. C.; Hohloch, S.; Lohrey, T. D.; Zarkesh, R. A.; Arnold, J.;* Anstey, M. R.* Dalton Transactions 2016, 45, 18653–18660.
Distance Dependent Quenching and Gamma-Ray Spectroscopy in Tin-Loaded Polystyrene Scintillators
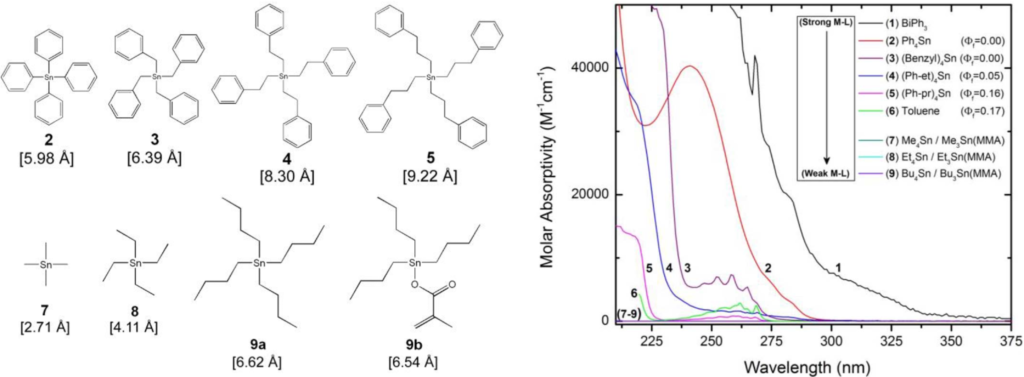
In this work, we report the synthesis and inclusion of rationally designed organotin compounds in polystyrene matrices as a route towards plastic scintillators capable of gamma-ray spectroscopy. Tin loading ratios of up to 15% w/w have been incorporated, resulting in photopeak energy resolution values as low as 10.9% for 662 keV gamma-rays. We also report fast scintillation decay behavior that is comparable to the quenched scintillators 0.5% trans-stilbene doped bibenzyl and the commercial plastic scintillator BC-422Q-1%.
- Feng, P. L.; Mengesha, W.; Anstey, M. R.; Cordaro, J. G. IEEE Transactions on Nuclear Science 2016, 63, 407–415.
Voltage Clustering in Redox-Active Ligand Complexes: Mitigating Electronic Communication Through Choice of Metal Ion
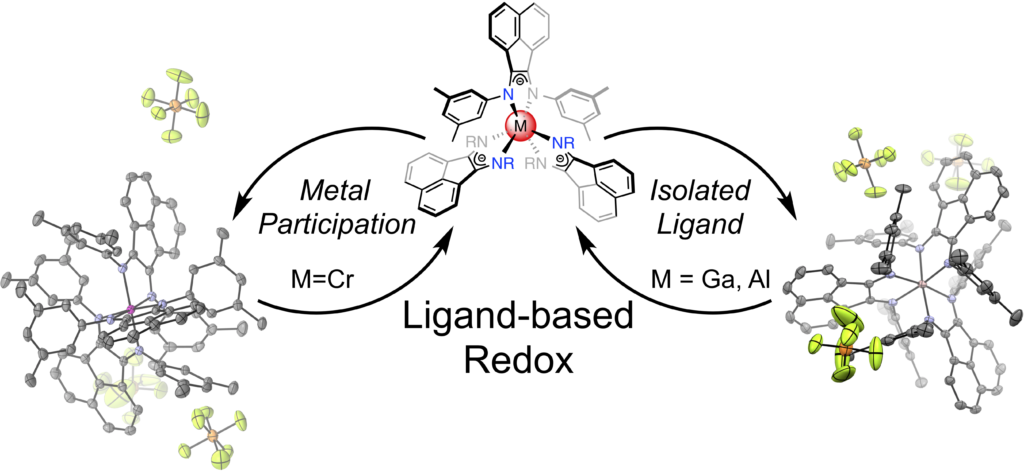
The redox-active bis(imino)acenapthene (BIAN) ligand was used to synthesize homoleptic aluminum, chromium, and gallium complexes of the general formula (BIAN)3M. The resulting compounds were characterized and modeled to compare the orbital contributions of main group elements and transition metals in ligand-based redox events. Complexes of this type have the potential to improve the energy density and electrolyte stability of grid-scale energy storage technologies, such as redox flow batteries, through thermodynamically-clustered redox events.
- Zarkesh, R. A.; Ichimura, A. S.; Monson, T. C.; Tomson, N. C.; Anstey, M. R.* Dalton Transactions 2016, 45 (24), 9962–9969.
- [Featured in a special issue entitled New Talent: Americas]
Redox Flow Batteries: An Engineering Perspective

Redox flow batteries are well suited to provide modular and scalable energy storage systems for a wide range of energy storage applications. In this paper, we review the development of redox-flow-battery technology including recent advances in new redox active materials, cell designs, and systems, all from the perspective of engineers interested in applying this technology. We discuss cost, performance, and reliability metrics that are critical for deployment of large flow-battery systems. The technology, while relatively young, has the potential for significant improvement through reduced materials costs, improved energy efficiency, and significant reduction in the overall system costs.
- Chalamala, B. R.; Soundappan, T.; Fisher, G. R.; Anstey, M. R.; Viswanathan, V. V.; Perry, M. L. Proceedings of the IEEE 2014, 102 (6), 976–999.
Application of Redox Non-Innocent Ligands to Non-Aqueous Flow Battery Electrolytes

High energy-density, redox flow batteries (RFB) can provide cost-effective, grid-scale energy storage, facilitating the use of intermittent sources such as solar and wind power. A new electrolyte based on vanadium and redox-active ligands that stores equivalents of charge separately from the metal center is presented. Electrolytes composed of non-innocent ligands greatly enhance both the energy density and stability of non-aqueous RFBs.
- Cappillino, P. J.; Pratt, H. D., III; Hudak, N. S.; Tomson, N. C.; Anderson, T. M.; Anstey, M. R.* Advanced Energy Materials 2014, No. 4, 1300566.
Lanthanide Corroles: A New Class of Macrocyclic Lanthanide Complexes

The first examples of lanthanide corroles are prepared by two synthetic routes. (Mes2(p-OMePh)corrole)La·4.5DME (1·4.5DME) and (Mes2(p-OMePh)corrole)Tb·4DME (2·4DME) are prepared from the free base corrole and Ln((NSiMe3)2)3, while (Mes2(p-OMePh)corrole)GdTACNMe3(3·TACNMe3) is prepared by metathesis of the recently reported Li3corrole and GdCl3.
- Buckley, H. L.; Anstey, M. R.; Gryko, D. T.; Arnold, J. Chemical Communications 2013, 49 (30), 3104–3106.
Surface Immobilization of an Organo-Iridium Complex Through a Carbon-Metal Bond

A new approach to the immobilization of organometallic complexes to monolayer surfaces is demonstrated using aldehyde-terminated self-assembled monolayers and a Cp*Ir complex. The iridium complex is anchored to the surface by direct attachment of the metal to the carbon chain of the monolayer film.
- Razgon, A.; Anstey, M. R.; Yakelis, N. A.; Bergman, R. G.; Sukenik, C. N. Inorganica Chimica Acta 2011, 375 (1), 305–307.
Ordered Metal Nanostructure Self-Assembly Using Metal-Organic Frameworks as Templates

We demonstrate that nanoporous metal–organic frameworks (MOFs) loaded with silver can serve as templates for ordered nanostructures comprising either silver nanoparticles or nanowires. Exposure to an electron beam breaks down the template, leading to rapid silver coalescence. The geometric and chemical structure of the MOF, as well as the extent of metal loading, determine whether nanoparticles or nanowires are formed and define their size and orientation. Nanowires with diameters as small as 4 nm and aspect ratios >125 can be formed, overcoming the limitations of existing templating methods. This method is relatively simple, compatible with many materials, and proceeds by a distinct template-directed growth mechanism. Since MOFs offer an unprecedented level of synthetic flexibility, combined with highly uniform porosity as a result of their crystalline structure, this approach opens a promising new route for synthesis of self-assembled, ordered nanostructures.
- Jacobs, B. W.; Houk, R. J. T.; Anstey, M. R.; House, S. D.; Robertson, I. M.; Talin, A. A.; Allendorf, M. D. Chemical Science 2011, 2 (3), 411-416.
Improved Synthesis of Bis(borano)hypophosphite Salts

A synthesis of the bis(borano)hypophosphite anion with various counterions has been developed to make use of more benign and commercially available reagents. This method avoids the use of potentially dangerous reagents used by previous methods and gives the final products in good yield. Details of the crystal structure determination of the sodium salt in space group Ama2 are given using a novel computational technique combined with Rietveld refinement.
- Anstey, M. R.; Corbett, M. T.; Majzoub, E. H.; Cordaro, J. G. Inorganic Chemistry 2010, 49, 8197–8199.
Photoinduced N2 Loss as a Route to Long-Lived Organometallic Alkane Complexes: A Time-Resolved IR and NMR Study
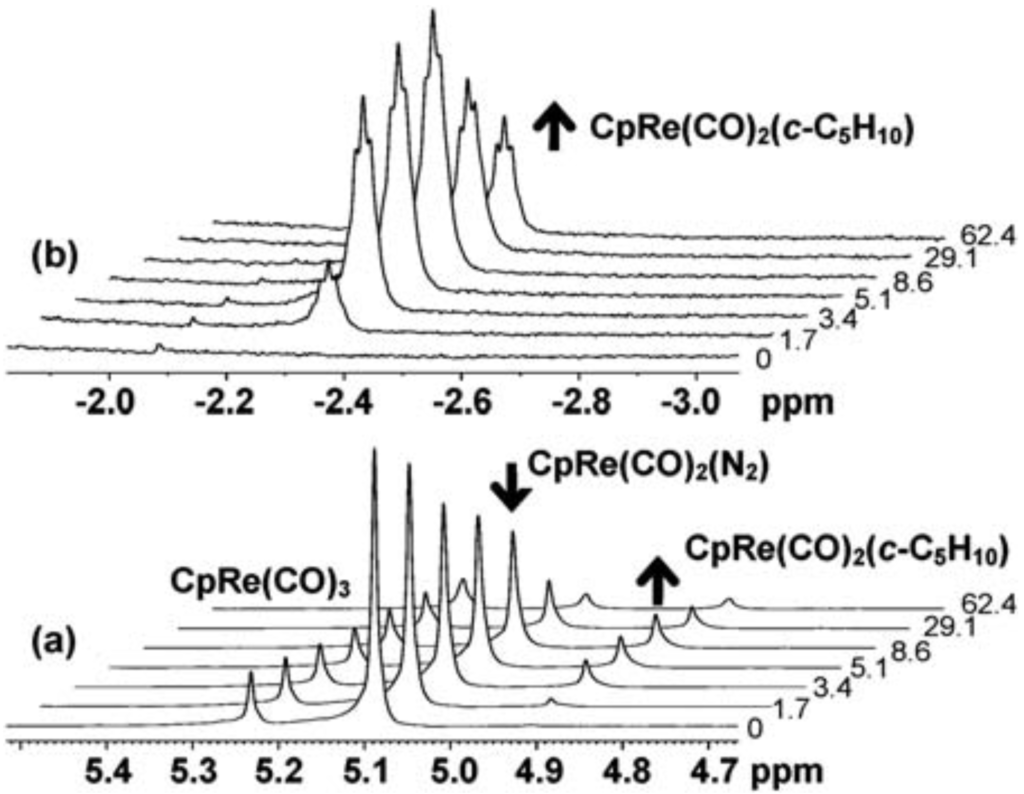
Photolysis of CpRe(CO)2(N2) and Cp‡Re(CO)2(N2) in alkane solutions with a UV lamp via a quartz fibre inserted into the NMR probe allows generation of a CpRe(CO)2(alkane) agostic complex. The major isomer of CpRe(CO)2(2,2-dimethylbutane-h2-C1,H1), exhibits a 1H NMR resonance for the co-ordinated hydrogen at δ = -2.19 with ¹J = 118 Hz. The photochemistry of Cp‡Re(CO)2(N2) (Cp‡ = h5-1,2-C5H3(tBu)2) in alkane solution is also reported. The reaction with cyclopentane has also been studied by NMR spectroscopy at 190 K with in situ laser irradiation at 355 nm. Cp‡Re(CO)2(C5H10) is shown to exhibit the characteristic features of an alkane complex in the NMR spectrum, viz. a large isotopic shift of the 1H resonance at δ = -2.44 upon partial deuteration of the alkane (Δδ = 1.77 ppm), a large ¹J (114 Hz) and a large negative 13C chemical shift (δ = -33.8).
- Calladine, J. A.; Torres, O.; Anstey, M.; Ball, G. E.; Bergman, R. G.; Curley, J.; Duckett, S. B.; George, M. W.; Gilson, A. I.; Lawes, D. J.; Perutz, R. N.; Sun, X.-Z.; Vollhardt, K. P. C. Chemical Science 2010, 1, 622–630.
Unexpected C-C Bond Cleavage and C-C Bond Formation Observed in the reaction of a Cationic Iridium Complex with Heteroatom-Substituted Cyclopropanes

Iridium complex Cp*(PMe3)Ir(CH3)OTf (1) and alkylaminocyclopropanes react via a well-understood initial C−H bond activation with a subsequent C−C bond-breaking step to yield a variety of π-allyl complexes [Cp*(PMe3)Ir(η3-C3H4NR1R2)][OTf] (R1 = R2 = Bn; R1 = Bn, R2 = Ph) and methane. However, diphenylamino, alkoxy, or siloxycyclopropanes, when treated with complex 1, yield a new π-allyl complex [Cp*(PMe3)Ir(η3-C3H4CH3)][OTf] (8) and the corresponding amine or alcohol. The methyl group initially bound to iridium is no longer extruded as methane, but instead is incorporated into the allyl moiety to give a new carbon−carbon bond. A detailed mechanistic study provides evidence in support of an initial C−C bond activation mechanism as opposed to the initial C−H bond activation observed with other known substrates.
- Anstey, M. R.; Yung, C. M.; Du, J.; Bergman, R. G. Journal of the American Chemical Society 2007, 129, 776–777.
Diastereo-and Enantioselective Dearomatization of Rhenium-bound Naphthalenes

Dihapto-coordinated naphthalene complexes of the form TpRe(CO)(L)(η²-naphthalene) (L = PMe3, pyridine, or 1-methylimidazole) undergo electrophilic addition with dimethoxymethane and with various Michael acceptors to generate 1H-naphthalenium species. These naphthalenium complexes undergo intra- or intermolecular nucleophilic addition reactions with stabilized enolates, silyl ketene acetals, or enols to form the corresponding dihydronaphthalene complexes. Oxidative decomplexation generates the free dihydronaphthalene. When a resolved form of the rhenium dearomatization agent is used, these reactions can be performed enantioselectively. DFT calculations provide a useful guide in explaining the observed stereochemistry. Depending on reaction conditions, a Michael-Michael ring-closure sequence (MIMIRC) or a net [2 + 4] cycloaddition with the bound naphthalene is also observed, and the corresponding tricyclic molecules can be removed from the metal in high yield.
Ding, F.; Valahovic, M. T.; Keane, J. M.; Anstey, M. R.; Sabat, M.; Trindle, C. O.; Harman, W. D. Journal of Organic Chemistry 2004, 69, 2257–2267.